|
Hongkai’s Research Group |

|
ChIP-seq, ChIP-chip, DNase-seq, ATAC-seq and other similar technologies are high-throughput methods for mapping genome-wide regulatory element activities. They are widely used to decode gene regulation in development and diseases. We develop analytical and software tools for analyzing these data. Examples of our tools include CisGenome, dPCA, TileMap, TileProbe, and JAMIE. We have also developed a database, hmChIP, to help scientists to explore publicly available ChIP data. |
|
2. Statistical and Computational Methods for Regulome Data (ChIP-seq, DNase-seq, ATAC-seq, etc.) |
|
Research |
|
6. Decoding Gene Regulation in Stem Cells, Development and Diseases |
|
We are interested in decoding gene regulatory programs in development, stem cells and diseases. We have contributed knowledge on gene regulation in (1) human and mouse embryonic stem cells [1,2], (2) the sonic hedgehog signaling pathway in embryonic development [3,4,5], (3) B cell lymphoma [1], leukemia [6], and various other cancers [7]. Most recently, we have started to develop systematic methods to predict genome-wide regulatory programs in development and diseases. |
|
7. Methods and Tools for New High-throughput Technologies |
|
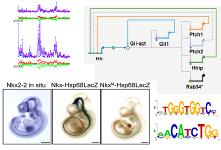
We also develop methods and tools for analyzing data from novel high-throughput technologies. One example is methods for analyzing TIP-chip data to identify active retrotransposon elements in human genomes. |
|
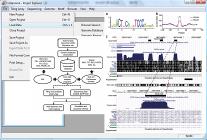
We develop tools for analyzing large scale gene expression data. One example is the correlation motif approach, CorMotif, for integrative analysis of multiple gene expression experiments. Most recently, we are interested in developing tools for mining the large amounts of gene expression data in gene expression omnibus (GEO). One example is Gene Set Context Analysis (GSCA), a method to help researchers systematically identify cell types, conditions and diseases associated with user-specified gene set activity patterns. |
|
3. Methods and Tools for Analyzing Large Scale Gene Expression Data |
|
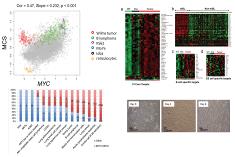
Integrative analysis of heterogeneous ‘omics datasets can lead to new discoveries. It can significantly increase the power for detecting weak signals or help one predict a system’s behavior. Data integration and data mining are non-trivial. Common issues include high dimensionality, heterogeneity, complex relationship among different data modalities, exponential complexity of combinatorial signal patterns, etc. We develop tools for data integration and mining that tackle these challenges. Examples include BIRD for predicting genome-wide regulatory element activities using gene expression, iASeq for integrative analysis of allele-specificity, JAMIE for joint analysis of multiple ChIP-chip datasets, CorMotif for joint analysis of multiple gene expression datasets, ChIP-PED for joint analysis of ChIP and public gene expression data. |
|
5. Statistical Methods for Data Integration and Data Mining |
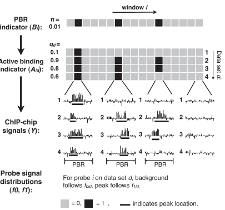
|
4. Methods and Tools for DNA Sequence Motif Analysis |

|
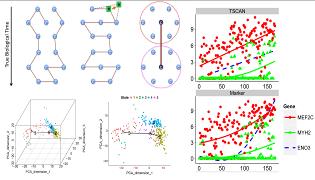
Single-cell genomic technologies such as single-cell RNA-seq, single-cell ATAC-seq and single-cell ChIP-seq provide unprecedented power for examining the functional genomic landscape of a heterogeneous cell population. We develop statistical and computational methods and tools for designing single-cell genomic experiments and analyzing single-cell genomic data. Examples of our tools include TSCAN and SCRAT. |
|
1. Analytical Methods and Tools for Single Cell Genomics |